

By Vincent Montoya & Jeffrey Joy
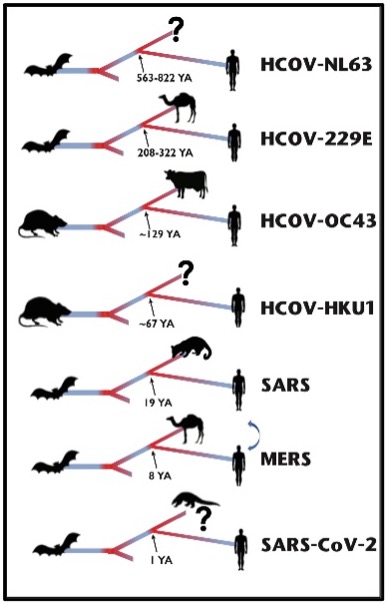
Last month we and our co-authors published our paper “Variable routes to genomic and host adaptation among coronaviruses” in the Journal of Evolutionary Biology. Coronaviruses are known to have spilled over into the human population on at least seven different occasions. In our study we compare the genomic adaptations of each of these seven coronaviruses to identify patterns and distinguishing characteristics along their evolutionary paths to their human hosts.
Viruses are the most ubiquitous biological entity on Earth. They exhibit rates of mutations that are often several orders of magnitude greater than their hosts and as a result are exquisitely adapted to these hostile intra/extracellular environments. This accelerated evolution is revealed on a daily basis through antiviral drug resistance, persistent immune evasion, rapid changes in host cell receptor binding affinities, and finally host switching events.
Host switching events offer unique insights into viral evolution where this accelerated evolution is stringently tested for its ability to adapt to the new cellular environment. The amount of viral adaptation necessary to establish endemic disease in the novel host is thought to be dependent upon the extent of genetic overlap between the donor and recipient hosts along with the frequency of contact between them [1]. Furthermore, the greater the evolutionary distance between hosts, the greater adaptation is required by the virus to overcome barriers to infection.
New host environments place enormous selective pressures on viruses to optimize host infection. Selection can be neutral where no changes were selected, positive which result in an increase in frequency of a particular mutation, or negative when a mutation at a particular site decreases in frequency or is purged (ie purifying selection).
Coronaviruses are known to have spilt over into the human population on at least seven different occasions [2]. These range from HCOV-NL63 which is thought to have diverged from its most recent common ancestor >500 years ago to SARS-CoV‑2 that is thought to have emerged in October 2019 [3] (Figure 1). A clear understanding of where, when, why, and how changes in the coronavirus genomes occur following a host jump is crucial for understanding new variants of the virus, vaccine design, public health responses, and predicting future pandemics. In this study we wanted to compare the genomic adaptations of each of these seven coronaviruses in order to identify underlying patterns and distinguishing characteristics along their evolutionary paths to their human hosts.
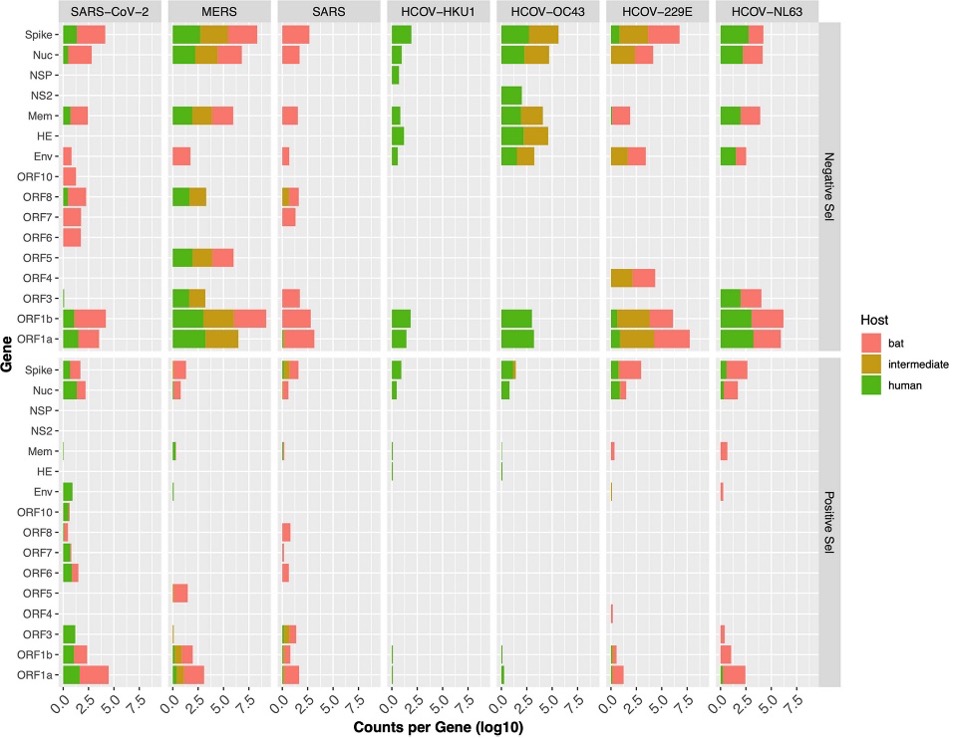
In order to examine the evolutionary selective pressures placed upon each virus separately for each host, all available genomes with host annotations were analyzed. First, we wanted to see if there were patterns in terms of which portion of the genome was under selection. We found that in general the genes associated with viral replication and the gene encoding the protein involved in host cell receptor binding (the Spike protein) were most commonly selected among each virus (Figure 2).
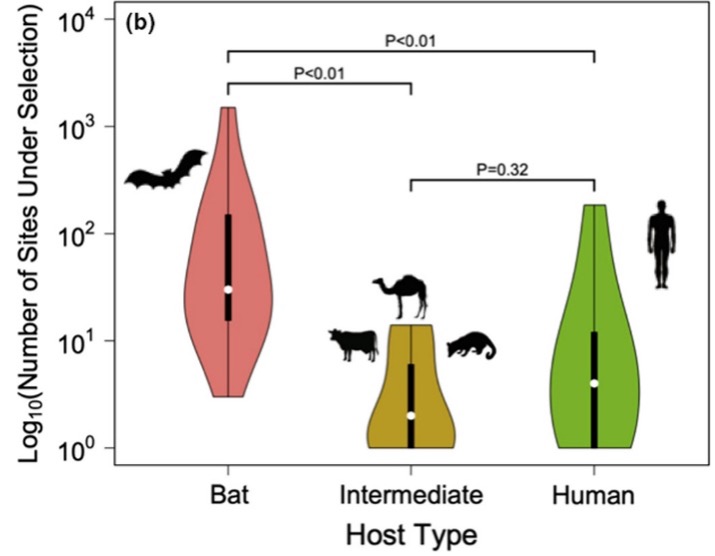
Next, we wanted to compare selective pressures by host type (Figure 3). We found that bats had significantly greater amounts of selection in the Spike and replication associated genes compared with human and intermediate hosts (civets, camels, and cows).
Finally, structural features of the Spike protein were examined to see whether they influenced the sites of selection for each of the coronaviruses. As the Spike protein is synthesized and subsequently shuttled to the cell surface, oligosaccharides are added and trimmed by host enzymes in a site-specific manner. One of the most common forms of these post-translational modifications is N‑linked glycosylation. For example, the SARS-CoV-2 trimeric spike protein is glycosylated at 22 different amino acid sites which collectively shield ~40% of the protein surface (Figure 4, [4]). These glycan molecules have significant impacts on several aspects of viral infections including protein stability, antigenicity, and host cell receptor binding.
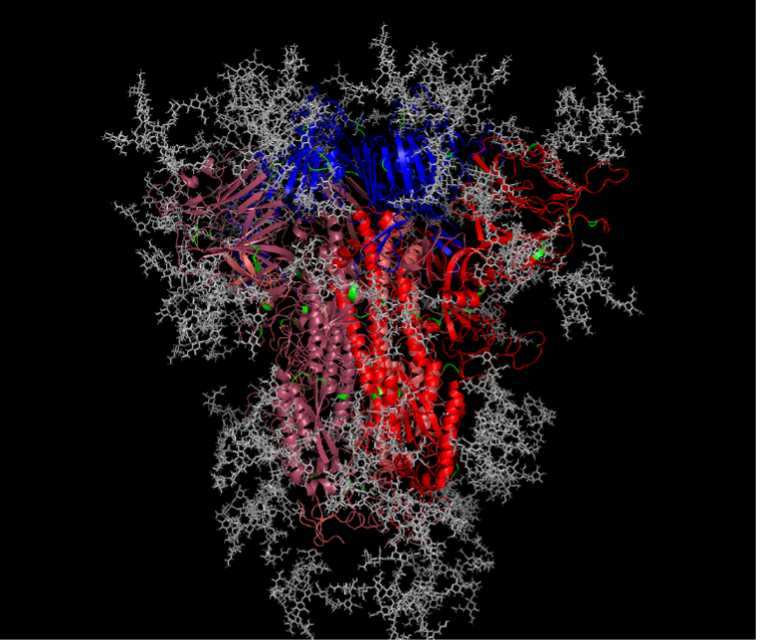
We sought to determine how these glycan molecules influence the evolutionary selective pressures placed upon each coronavirus. To address this question, we compared the proximity of each glycan molecule to each site that was under positive selection. If sites under positive selection were more commonly found at a greater distance relative to glycan molecules, then it suggests that glycans are shielding or blocking selection (for example, antibody driven evolution). Consistent with previous results we found these distances varied, however, a consistently higher fraction of sites under positive selection were in close proximity to glycan molecules for SARS-CoV‑2. This suggests that selection, while multifactorial, is perhaps less driven by antibody evasion and to a greater extent by protein stabilization and/or host receptor binding.
These results support the hypothesis that each coronavirus has traversed a different path along the way to colonizing humans and highlight the extraordinary capacity of evolution to solve problems in a myriad of ways. The selective regimes placed upon the genomes of each virus seems to reflect the delicate balance between viral genetics and the unique ecologies of both the viruses and their respective hosts.
References
[1] K. J. Olival, P. R. Hosseini, C. Zambrana-Torrelio, N. Ross, T. L. Bogich, and P. Daszak. (2017). Host and viral traits predict zoonotic spillover from mammals, Nature, 546, 646– 650, doi: 10.1038/nature22975.
[2] J. Cui, F. Li, and Z. L. Shi. (2019). Origin and evolution of pathogenic coronaviruses, Nature Reviews Microbiology. 2019, 17, 181–192, doi: 10.1038/s41579-018‑0118‑9.
[3] J. Pekar, M. Worobey, N. Moshiri, K. Scheffler, and J. Wertheim. (2021). Timing the SARS-CoV‑2 index case in Hubei province. Science, 372, 412–417, doi: 10.1126/science.abf8003.
[4] Y. Watanabe, J. D. Allen, D. Wrapp, J. S. McLellan, and M. Crispin. (2020). Site-specific glycan analysis of the SARS-CoV‑2 spike, Science, 369, 330– 333, doi: 10.1126/science.abb9983.

